Intermetallische Kooperativität in supramolekularen Komplexverbindungen
Assoziierte Arbeitsgruppe Prof. Wolfram Seidel (Universität Rostock)
Die Forschungsgruppe beschäftigt sich grundsätzlich mit koordinationschemischen und metallorganischen Fragestellungen, wobei die klassische Synthesechemie zu den Kernkompetenzen der Gruppe zählt. Im Zentrum des Interesses stehen dabei kooperative Effekte zwischen verschiedenartigen Metallzentren, die mit möglichst hoher Effektivität über spezifische Liganden vermittelt werden. Kooperative Wechselwirkungen bilden nicht nur die Grundlage für moderne, intelligente Materialien wie Einzelmolekülmagnete oder Substanzen für die Umwandlung von Licht in speicherfähige Energieformen, auch die Wirkungsweise einer großen Zahl von Metalloproteinen basiert auf bimetallischen Reaktionsmustern. In derartigen Systemen mit mehreren kooperativ wirkenden Metallzentren können reaktionsträge Substrate aktiviert oder lichtgetriebene Ladungstrennungen als zentralem Vorgang jeder Photokatalyse erzwungen werden. Unsere Forschungsarbeiten lassen sich dabei in folgende zwei Themenbereiche einteilen:
1. Untersuchungen zum koordinationschemischen Potential von Alkinliganden, die in beiden α-Positionen über Heteroatome verfügen
2. Entwicklung neuartiger Dithiolenliganden als funktionale Brückenliganden

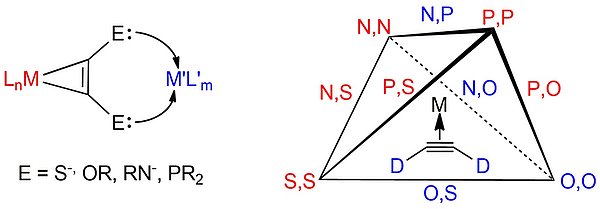
Als Donorzentren für Alkinliganden kommen neben terminalem Sauerstoff -O– und Stickstoff -NR– vor allem klassische Donoren der 3. Periode wie Schwefel -S– und Phosphor als -PR2 in Betracht. Von herausragendem Interesse sind dabei neben der Entwicklung neuer Koordinationsmotive bzw. Konnektivitäten umfassende Untersuchungen zur elektronischen Struktur und zum kooperativen Verhalten der verbundenen Metallzentren. Die schematische Darstellung möglicher Donoratom-Kombinationen (siehe Abb.) führt bei der Beschränkung auf die Elemente Sauerstoff, Schwefel, Stickstoff und Phosphor zu einem Tetraeder, dessen Kanten die Kombination unterschiedlicher Donoratome am Alkin repräsentieren. Von allen möglichen sind die rot markierten Kombinationen von uns in den letzten Jahren realisiert worden.
Die Motivation für diese Untersuchungen liegt einerseits in der grundsätzlichen Frage nach den elektronischen Wechselwirkungen der verknüpften Metalle. In Abhängigkeit von der Elektronenkonfiguration der Metalle und der Hybridisierung der Donorzentren werden entweder delokalisierte oder lokalisierte Zustände gefunden. Ersteres führt bei verbreiteter Redoxaktivität der mehrkernigen Komplexverbindungen zum Phänomen der Gemischtvalenz, während wir für lokalisierte Zustände echte Redoxisomerie gefunden haben. Die anwendungsbezogenen Aspekte des Ansatzes liegen in der Entwicklung redoxaktiver Metalloliganden für die Katalyse. Ein potenziell redox-aktiver Alkinkomplex kann als Ligand mit Metallen der ersten Übergangsmetallreihe, die bei normalem Potenzial Ein-Elektronen-Redoxprozesse wie Fe(II)/Fe(III) oder Cu(I)/Cu(II) durchlaufen, die Basisreaktionen oxidative Addition/reduktive Eliminierung fördern, indem das zweite Redoxäquivalent durch den Alkinkomplex-Liganden zur Verfügung steht. Neben der Einstellung geeigneter elektrochemischer Potenziale ist dafür die elektronische Kooperativität der Metalle von großer Bedeutung.
Für diese Anwendungen suchen wir optimale Kombinationen von Metallionen und Komplexbausteinen, die zu erstrebenswerten Eigenschaften führen wie gute Löslichkeit, Konfigurationsstabilität, Redoxvariabilität in ähnlichen Potenzialfenstern, hohe Absorptivität und nicht zuletzt das Vorhandensein guter spektroskopischer Sonden wie CO-Liganden für IR-Spektroskopie oder Metalle mit geeignetem Kernspin für die EPR-Spektroskopie. Letzteres ist fundamental für die Aufklärung der elektronischen Strukturen in unterschiedlichen Redoxzuständen. Die zugehörigen Untersuchungen schließen deshalb neben der umfassenden Routine-Charakterisierung (Röntgenbeugung, Multikern-NMR-Spektroskopie, IR/Raman-Spektroskopie) speziellere Methoden wie EPR-Spektroskopie, Zyklovoltammetrie, NIR-, UV-vis- und Fluoreszenz-Spektroskopie sowie Spektroelektrochemie ein.
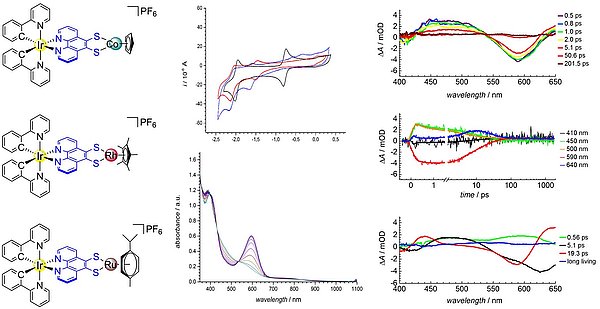
Die Arbeiten zum Thema lichtinduzierte Ladungstrennung und Energieübertragung in polynuklearen Komplexen basieren auf Untersuchungen mit Phenanthrolin-5,6-dithiolat (phendt2‒). Phenanthroline mit Donor-Substitution in der 5,6-Position sind besonders starre Brückenliganden, die eine vorteilhafte Bedingung für sehr schnelle Elektronentransferprozesse zwischen verbundenen Metallen erfüllen. Im Jahr 2014 stellten wir die ersten dinuklearen Komplexe mit phendt2‒ vor, die N,N'-gebundene Ru(bpy)2- und Ir(ppy)2-Photozentren mit dithiolat-koordinierten zweiwertigen Metallen der Gruppe 10 kombinierten. Darauf aufbauende Untersuchungen zielten auf die Einstellung eines thermodynamisch günstigen Redoxpotenzials der Metalle an der Dithiolateinheit für einen photoinduzierten, gerichteten Elektronentransfer. In allen Fällen bewirkt die Koordination eines zweiten Metalls am Dithiolat-Chelat ein wirksames Photolumineszenz-Quenching. Seit 2014 existiert eine enge Zusammenarbeit mit der Arbeitsgruppe von Stefan Lochbrunner am Fachbereich Physik der Universität Rostock, die die zeitaufgelöste spektroskopische Charakterisierung der Komplexe durchgeführt. Für die Verbindung [Ir(ppy)2(µ-phendt)Co(C5H5)] konnten wir mittels fs-transienter Absorptionsspektroskopie einen ultraschnellen Dexter-Energietransfer vom Ir-Komplex auf die CpCo-Einheit nachweisen, wobei allerdings nicht von einem konsekutiven Elektronen- und Lochtransfermechanismus unterschieden werden konnte (siehe Abb.). Folglich sind verlängerte Phenanthrolinliganden ideale Plattformen für ultraschnelle Elektronentransferprozesse.
Weitere Informationen finden Sie auf unserer Uni-Hompage.
Literatur:
Migratory insertion of isocyanide into a ketenyl–tungsten bond as key step in cyclization reactions C. Timmermann, P. Thiem, D. Wanitschke, M. Hüttenschmidt, J. Romischke, A. Villinger, W. W. Seidel, Chem. Sci. 2021, 10.1039/D1SC06149F
Metal/Metal Redox Isomerism Governed by Configuration S. Ludwig, K. Helmdach, M. Hüttenschmidt, E. Oberem, J. Rabeah, A. Villinger, R. Ludwig, W. W. Seidel, Chem. Eur. J. 2020, 26, 16811-16817.
Facile Synthesis of a stable side-on Phosphinyne Complex by Redox Driven Intramolecular Cyclisation H. Lange, H. Schröder, E. Oberem, A. Villinger, J. Rabeah, R. Ludwig, K. Neymeyr, W. W. Seidel, Chem. Eur. J. 2020, 26, 11492-11502.
Pathways to Polynuclear Complexes with α-S,N-Substituted Alkynes as Bridging Ligand J Rüger, C. Timmermann, A. Villinger, W. W. Seidel, Inorg. Chem. 2019, 58, 9270-9279.
1,10-Phenanthroline-dithiine iridium and ruthenium complexes: Synthesis, characterization and photocatalytic dihydrogen evolution E. Erdmann, A. Villinger, B. König, W. W. Seidel, Photochem. Photobiol. Sci. 2018, 17, 1056–1067.
Ultrafast Energy Transfer in Dinuclear Complexes with Bridging 1,10-Phenanthroline-5,6-Dithiolate E. Erdmann, M. Lütgens, S. Lochbrunner, W. W. Seidel, Inorg. Chem. 2018, 57, 4849–4863.
Sterically encumbered metalla-diphosphines: unlocking alkyne rotation by PtII coordination K. Helmdach, S. Dörk, A. Villinger, W. W. Seidel, Dalton Trans. 2017, 46, 11140-11144.
Synthesis and activation potential of an open shell diphosphine K. Helmdach, S. Ludwig, A. Villinger, D. Hollmann, J. Kösters, W. W. Seidel, Chem. Commun. 2017, 53, 5894–5897.
Minimalistic Ditopic Ligands: An α-S,N-Donor-Substituted Alkyne as Effective Intermetallic Conjugation Linker J. Rüger, C. Timmermann, A. Villinger, A. Hinz, D. Hollmann, W. W. Seidel, Chem. Eur. J. 2016, 22, 11191–11195.
Dual nucleophilic substitution at a W(II) η2-coordinated diiodo acetylene leading to an amidinium carbyne complex K. Helmdach, J. Rüger, A. Villinger, W. W. Seidel, Chem. Commun. 2016, 52, 2616–2619.