Catalytic Cycloadditions
Associated Group Prof. Marko Hapke (University Linz, Austria)
The liaison of unsaturated substrates containing C-C double or triple bonds and transition metal complexes provides the entry into a fascinating world of unusual reactions and the formation of a wide spectrum of different reaction products. The construction of molecules with high functionality and molecular complexity by transition metal-catalysed cycloaddition reactions, starting from alkynes and heteroatom-containing analogues, constitutes a very multi-faceted challenge. Therefore our particular attention is dedicated to the systematic praparation and characterisation of novel precatalysts based on group 9 metals and neighbouring metals like iron or nickel. Especially half-sandwich cobalt compounds and from them derived complexes can contribute significantly by studying their reactivity and underlying structural parameters.[1] Insightful findings for the catalytic assembly of benzenes and heteroarenes by cyclotrimerization reactions were gained by comparison with the corresponding complexes of the heavier homologues rhodium and iridium as catalysts.[2]
Additionally a major role is played by the task of investigating the cobalt-catalysed asymmetric [2+2+2] cycloaddition for the enantioselective preparation of axially chiral biaryl derivatives. The photochemical conditions applied contrast the pure thermal reaction with exceptional results and observations. Due to the necessary preparation of the required substrates needed for the investigation of novel precatalysts, we have acquired extensive knowlege in the area of preparative alkyne and arene chemistry.
Cyclopentadienyl (Cp) cobalt(I) complexes rank among the "1st generation" catalysts for [2+2+2] cycloaddition reactions; however, only a small number of complexes are currently applied in practice and most of them require relatively high activation temperatures. We have synthesised an array of new cobalt(I)-olefin complexes and evaluated their high reactivity in cycloaddition reactions.[3] The introduction of phosphites as ligands and the development of new synthetic methods led to a number of catalysts, which are active already at very mild temperatures and provide scope for systematic studies of the reactivity, depending on the ligand combination.[4]
Evaluation of novel CpCo-phosphite complexes in cycloaddition reactions:
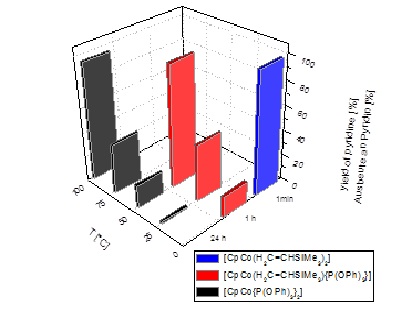
Furthermore we have prepared, for the first time, complexes with an identical organic ligand framework for all metals of group 9; this made it possible to compare reactivity and selectivity in cyclotrimerisations, hydrogenations and hydroformylations based on the intrinsic differences of the metals.[5] For a highly reactive CpCo(I)-complex we confirmed the formation of a tetranuclear Co4-cluster complex in non-chlorinated solvents at low temperatures and were able to isolate and completely characterize the cluster;[6] this cluster exhibited interesting properties in isomerization reactions of terminal olefins.
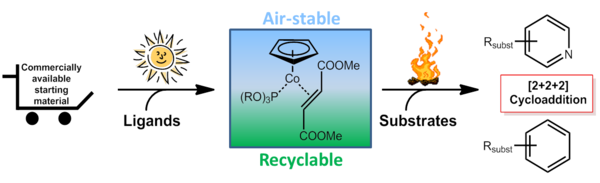
The further development of mixed CpCo-olefin-phosphite complexes led to the design of rare examples for air-stable precatalysts, which can be applied under thermal as well as photochemical reaction conditions.[7] This strategy for the stabilisation of Co(I)-complexes containing different donor ligands was successfully passed on to complexes possessing functionalised Cp rings, which were investigated for the connection to solid phase materials.[8]
Air stable and recyclable CpCo(l) precatalysts:
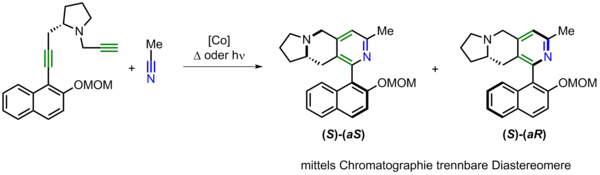
The stereoselective synthesis of axially chiral biaryls (two aryl rings connected by a single bond, which cannot rotate freely) presents a highly important topic in organic synthesis. Biaryls containing axial chirality as the stereogenic element play an increasingly important role in natural products as well as in pharmaceutical ingredients, while being long established ligands in catalytic applications. We have investigated in detail the application of chiral Co(I)-complexes under photochemical conditions for the asymmetric synthesis of heterobiaryls.[9] Furthermore the cobalt-catalyzed synthesis of diastereomeric atropisomers starting from chiral diynes has been demonstrated.[10] The study confirmed the approach adopted – that of not only preparing both diastereomers in one reaction but also of subsequently separating them easily by chromatography, leading to the pure atropisomers. The reaction also made possible the direct comparison of thermal and photochemical reaction conditions in a practical application.
Darstellung von trennbaren Diastereomeren:
We also applied the cycloaddition reaction for the assembly of biaryl systems containing five-membered ring systems in the backbone of the pyridine moiety of the molecule, thus enabling the rotational barrier of the biaryl axis to be investigated. These investigations demonstrated the exceptional dependence of the stability of the biaryl axis on small structural variations. The synthesis of different symmetrically and unsymmetrically substituted triynes resulted in the construction of a large variety of novel, functionalised triaryls, including the identification of CoCl(PPh3)3 as a highly suitable cyclotrimerization catalyst under partly very mild conditions.[11] The results provided evidence for the high suitability of the cobalt-catalysed [2+2+2] cycloaddition reaction for the synthesis of a wide range of different biaryls.
Our expertise with reactions that include gaseous acetylene occupies another important role in the field of transition metal-catalyzed hydroheterofunctionalization. These reactions make use of the potential of unsaturated compounds and especially acetylene for functionalization particularly by utilizing late transition metal complexes, e.g. Ni, Pd, Pt and Fe. These functionalization reactions are especially appealing, because they produce very useful building blocks with high functionality from relatively simple precursor molecules.
References
- I. Thiel, M. Hapke, Rev. Inorg. Chem. 2014, 34, 217-245.
- B. Heller, M. Hapke, Chem. Soc. Rev. 2007, 36, 1085-1094.
- M. Hapke, N. Weding, A. Spannenberg, Organometallics 2010, 29, 4298-4304.
- I. Thiel, H. Jiao, A. Spannenberg, M. Hapke, Chem. Eur. J. 2013, 19, 2548-2554.
- a) N. Weding, R. Jackstell, H. Jiao, A. Spannenberg, M. Hapke, Adv. Synth. Catal. 2011, 353, 3423-3433; b) Übersicht: N. Weding, M. Hapke, Chem. Soc. Rev. 2011, 40, 4525-4538.
- N. Weding, A. Spannenberg, R. Jackstell, M. Hapke, Organometallics 2012, 31, 5660-5663.
- I. Thiel, A. Spannenberg, M. Hapke, ChemCatChem 2013, 5, 2865-2868.
- I. Thiel, M. Hapke, J. Mol. Catal. A: Chem. 2014, 383-384, 153-158.
- a) A. Gutnov, B. Heller, C. Fischer, H.-J. Drexler, A. Spannenberg, B. Sundermann, C. Sundermann, Angew. Chem. 2004, 116, 3883-3886; b) M. Hapke, K. Kral, C. Fischer, A. Spannenberg, A. Gutnov, D. Redkin, B. Heller, J. Org. Chem. 2010, 75, 3993-4003.
- F. Fischer, P. Jungk, N. Weding, A. Spannenberg, H. Ott, M. Hapke, Eur. J. Org. Chem. 2012, 5828-5838.
- P. Jungk, F. Fischer, I. Thiel, M. Hapke, J. Org. Chem. 2015, 80, 9781-9793.
