Elektrochemie & Katalyse
Prof. Robert Francke
Die effiziente Nutzung von Energie und Materialien sowie die Einführung erneuerbarer Ressourcen in Materialkreisläufe sind Kernziele einer Kreislaufwirtschaft. Die Elektrochemie entwickelt sich zu einer Schlüsseltechnologie zur Erreichung dieser Ziele. Zum einen wächst der Anteil grüner elektrischer Energie aus Windkraft und Photovoltaik. Zum anderen gelingt mit Hilfe der Elektrosynthese die Substitution chemischer Oxidations- bzw. Reduktionsmittel durch grüne elektrische Energie als treibende Kraft chemischer Reaktionen.
Eine der großen Herausforderungen in der Elektrosynthese ist die kinetische Hemmung des Elektronentransfers, die oft zu Selektivitätsproblemen und erhöhtem Energieverbrauch ("Überspannung") führt. Vor diesem Hintergrund fokussieren wir unsere Arbeiten auf die Entwicklung homogener und heterogener elektrokatalytischer Systeme für die Umsetzung organischer Verbindungen und kleiner Moleküle wie CO2, H2O und N2. Die Nutzung substöchiometrischer Ladungsmengen für die Katalyse redoxneutraler Reaktionen („elektrochemische Katalyse“) bietet einen weiteren vielversprechenden und bisher wenig untersuchten Ansatz, den wir derzeit im Rahmen von molekularen Umlagerungen untersuchen. Um eine wissensbasierte Optimierung unserer Reaktionen zu ermöglichen, ist das mechanistische Verständnis des katalytischen Prozesses für uns von besonderem Interesse. Hierfür setzen wir elektroanalytische Techniken ein und kombinieren diese gegebenenfalls mit spektroskopischen Methoden und Kontrollexperimenten.
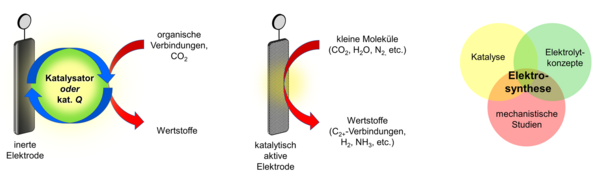
Ein weiteres Problem ist die Notwendigkeit zur Verwendung großer Leitsalzmengen. Nach Abschluss der Elektrolyse muss das Salz aus dem Reaktionsgemisch abgetrennt werden. Da das Salzadditiv sowohl eine potenzielle Abfallquelle als auch einen erheblichen Kostenfaktor darstellt, ist ein Recycling wünschenswert. In ähnlicher Weise stellen auch molekulare Katalysatoren ein Trenn- und Abfallproblem dar. In diesem Zusammenhang setzen wir einen weiteren Schwerpunkt auf das Thema „Multifunktionale Elektrolyte“.
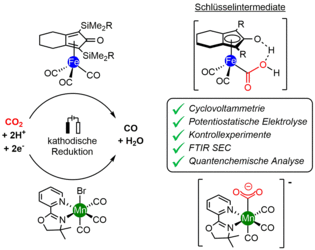
In den vergangenen Jahren haben wir ein Forschungsprogramm entwickelt, das sich auf die Nutzung von metallorganischen Katalysatoren für die Elektrosynthese fokussiert [1-5]. In einer Studie wurde ein System auf der Basis von Cyclopentadienon-Eisenkomplexen untersucht, [1,2] das eine Umsetzung von CO2 zu CO in nahezu quantitativer Faraday-Ausbeute und hoher Stromdichte ermöglicht. Mit einer Kombination aus zyklischer Voltammetrie, potentiostatischer Elektrolyse, FTIR-Spektroelektrochemie (in Zusammenarbeit mit der Gruppe Schwingungsspektroskopie in der Katalyse) und quantenchemischen Modellierungen (in Zusammenarbeit mit der Gruppe von Dr. Michael Römelt, HU Berlin) konnten wir mechanistische Erkenntnisse gewinnen und eine Sequenz vorschlagen, die über das in der Abbildung oben rechts gezeigte Schlüsselintermediat verläuft. In einem weiteren Projekt haben wir ein Mangan-Diimin-Komplex unter elektro- und photochemischen Bedingungen untersucht (in Zusammenarbeit mit der Gruppe Katalyse für Energietechnologien) .[3]
Referenzen:
[1] E. Oberem, A. F. Roesel, A. Rosas-Hérnandez, T. Kull, S. Fischer, A. Spannenberg, H. Junge, M. Beller, R. Ludwig, M. Roemelt, R. Francke, Organometallics 2019, 38, 1236–1247.
[2] A. Rosas-Hernández, H. Junge, M. Beller, M. Roemelt, R. Francke, Catal. Sci. Technol. 2017, 7, 459−465.
[3] C. Steinlechner, A. F. Roesel, E. Oberem, A. Paepcke, N. Rockstroh, F. Gloaguen, S. Lochbrunner, R. Ludwig, A. Spannenberg, H. Junge, R. Francke, M. Beller, ACS Catal. 2019, 9, 2091−2100.
[4] R. Francke, M. Roemelt, B. Schille, Chem. Rev. 2018, 118, 4631–4701.
[5] H. Liang, T. Beweries, R. Francke, M. Beller, Angew. Chem. Int. Ed. 2022, e202200723.
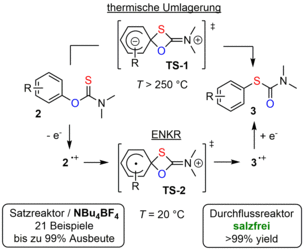
Im Vergleich zur Elektrokatalyse, bei der eine chemische Wechselwirkung zwischen einem Edukt und einem Katalysator den Elektrodenprozess erleichtert, stellt die elektrochemische Katalyse genau den umgekehrten Fall dar. Hier katalysiert die Übertragung einer positiven oder negativen Ladung auf ein Edukt unter milden Bedingungen eine redoxneutrale Reaktion, die andernfalls harsche Bedingungen und/oder den Einsatz von Reagenzien erfordern würde [1]. Bei solchen Reaktionen wird zunächst eine ionische oder radikalionische Spezies generiert, die nach einem gekoppelten chemischen Schritt entweder einen rückwärts gerichteten Elektronenaustausch mit der Elektrode durchläuft (ECEb-Mechanismus) oder einen Kettenprozess im Lösungsinneren anstößt. Unter diesen Umständen reichen substöchiometrische Ladungsmengen aus, um einen vollständigen Umsatz zu erreichen. Konzeptionell können somit die injizierten Elektronen und Löcher als Katalysatoren aufgefasst werden. In diesem Zusammenhang beschäftigen wir uns mit redoxneutralen Reaktionen wie Umlagerungen und Cycloadditionen. In einem unserer Projekte haben wir eine elektrochemisch katalysierte Variante der Newman-Kwart-Reaktion (ENKR) von O-Arylthiocarbamaten zu den entsprechenden S-Arylverbindungen entwickelt (siehe Abbildung). Im Allgemeinen stellt die NKR die Schlüsselreaktion in der dreistufigen Synthese von Thiophenolen aus deren Phenolderivaten dar und verläuft zwischen 200 und 300 °C. Die elektrochemische Katalyse ermöglicht hingegen einen Betrieb bei Raumtemperatur und bietet eine komplementäre Reaktivität in Bezug auf die Arylsubstitution [2,3]. Die Durchführung im Mikrodurchflussreaktor erlaubt es sogar, gänzlich auf das Leitsalz zu verzichten.
Referenzen:
[1] R. Francke, R. D. Little, ChemElectroChem. 2019, 6, 4373.
[2] T. Broese, A. F. Roesel, A. Prudlik, R. Francke, Org. Lett. 2018, 20, 7483.
[3] A. F. Roesel, M. Ugandi, N. T. T. Huyen, M. Májek, T. Broese, M. Roemelt, R. Francke, J. Org. Chem. 2020, 85, 8029-8044.
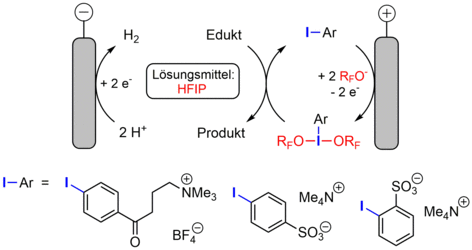
In den vergangenen fünf Jahren haben wir mehrere Ansätze zur Vereinfachung der Aufarbeitung von indirekten Elektrosynthesen untersucht. Einer dieser Ansätze basiert auf der Zusammenführung von Redox-Mediator und Leitsalz in eine einzige molekular definierte Spezies (siehe Abbildung, links). Aufgrund des breiten Anwendungsspektrums von hypervalenten Iod-Reagenzien in der organischen Synthese [1] haben wir uns dazu entschieden, das Iod(I)/Iod(III)-Paar als Studienobjekt zu verwenden. Eine Reihe von Untersuchungen an ionischen Aryliodiden zeigte, dass bei bestimmten Bedingungen die Elektrolyse im Satzreaktor ohne den Einsatz von Salzadditiven möglich ist und die Abtrennung und Wiederverwendung des Mediators deutlich erleichtert wird [2-4]. Unter Verwendung von 1,1,1,3,3,3-Hexafluorisopropanol (HFIP) als Lösungsmittel fanden wir heraus, dass die aktiven Dialkoxy-λ3-Iodane in nahezu quantitativen Faraday-Ausbeuten gebildet werden (siehe Abbildung). Die Post-Elektrolyselösungen können als reaktive Medien für eine Reihe von oxidativen C-O-, C-C- und C-N-Kupplungen eingesetzt werden. Der elektrochemische Ansatz konnte mittlerweile auch auf hypervalente Bromverbindungen übertragen werden [5].
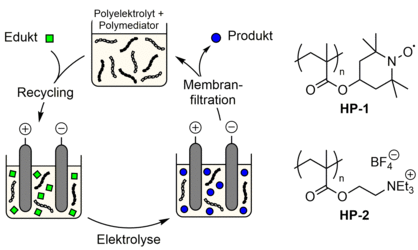
Ein weiterer Ansatz basiert darauf, Katalysatormoleküle und Leitsalze an lösliche Polymere zu binden ("Polymediatoren und Polyelektrolyte"), was eine Trennung beider Komponenten in einem einzigen Schritt unter Verwendung von Größenausschlussmembranen ermöglicht (siehe Abbildung). Am Beispiel der TEMPO-katalysierten Alkoholoxidation haben wir erstmals gezeigt, dass die indirekte Elektrosynthese unter Verwendung des Polyelektrolyten HP-1 und des Polymediators HP-2 auf effiziente Weise mit der Dialyse bzw. der Ultrafiltration gekoppelt werden kann [6,7] Die Polymerlösungen sind ausreichend leitfähig, zeigen eine hohe elektrokatalytische Aktivität gegenüber der Oxidation verschiedener Alkohole und können wiederverwendet werden.
Referenzen:
[1] R. Francke, Curr. Opin. Electrochem. 2021, 28, 100719.
[2] A. F. Roesel, T. Broese, M. Májek, R. Francke, ChemElectroChem. 2019, 6, 4229–4237.
[3] O. Koleda, T. Broese, J. Noetzel, M. Roemelt, E. Suna, R. Francke, J. Org. Chem. 2017, 82, 11669–11681.
[4] T. Broese, R. Francke, Org. Lett. 2016, 18, 5896–5899.
[5] I. Sokolovs, N. Mohebbati, R. Francke, E. Suna, Angew. Chem. Int. Ed. 2021, 60, 15832-15837.
[6] B. Schille, N. O. Giltzau, R. Francke, Angew. Chem. Int. Ed. 2018, 57, 422.
[7] N. Mohebbati, A. Prudlik, A. Scherkus, A. Gudkova, R. Francke, ChemElectroChem 2021, 8, 3837-3843.
Der Bereich "Elektrochemie & Katalyse" ist in zwei Themen untergliedert:
- Katalysatordesign für die Elektrosynthese (Leitung: Dr. Bernd Müller)
- Molekulare Elektrochemie (Leitung: Prof. Robert Francke)
- Kontinuierliche elektrochemische Prozesse (Leitung: Dr. Wen Ju)
sowie einer explorativen Themengruppe.
- Heterogene Elektrokatalysatoren (Leitung: Dr. Annette-Enrica Surkus)


