Intermetallic cooperativity in supramolecular coordination compounds
Associated Research Group Prof. Wolfram Seidel (University Rostock)
The research group is fundamentally concerned with coordination chemistry and organometallics, with synthesis being the core competence of the group. The focus of interest is on electronic cooperativity between different metals mediated via specific bridging ligands, which exhibit a high conjugation potential. Cooperative interactions not only form the basis for modern, intelligent materials such as single-molecule magnets or substances for the conversion of light into storable forms of energy, but the extraordinary activity of a large number of metallo-proteins is also based on bimetallic reaction patterns. In such systems with several cooperatively acting metal centers, inert substrates can be activated or light-driven charge separations can be forced as a principal process of any photocatalysis. Our research work can be divided into the following two topics:
1. Studies coordination chemical potential of alkyne ligands that bear heteroatoms in both α-positions
2. Development of novel dithiolene ligands as functional bridging ligands

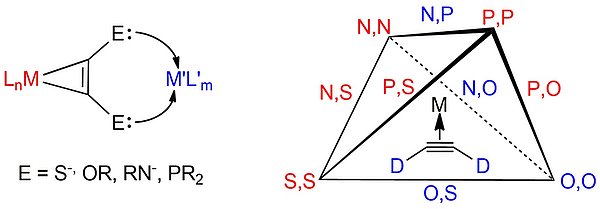
Besides terminal oxygen -O- and nitrogen -NR-, classical donors of the 3rd period such as sulfur -S- and phosphorus as -PR2 can be considered as donor centers for alkyne ligands. Of outstanding interest, besides the development of new coordination motifs or connectivities, are comprehensive investigations of the electronic structure and cooperative behavior of the linked metal centers. The schematic representation of possible donor atom combinations (see Figure), when restricted to the elements oxygen, sulfur, nitrogen and phosphorus, leads to a tetrahedron whose edges represent the combination of different donor atoms on the alkyne. Of all possible ones, the combinations marked in red have been realized by us in recent years.
The motivation for these investigations lies, on the one hand, in the fundamental question of the electronic interactions of the linked metals. Depending on the electron configuration of the metals and the hybridization of the donor centers, either delocalized or localized states are found. The former leads to the phenomenon of mixed valence in the presence of widespread redox activity of the multinuclear complex compounds, while we found true redox isomerism for localized states. The application-related aspects of the approach lie in the development of redox-active metalloligands for catalysis. A potentially redox-active alkyne complex can act as a ligand with metals of the first transition metal series undergoing one-electron redox processes such as Fe(II)/Fe(III) or Cu(I)/Cu(II) at normal potential to promote the basic oxidative addition/reductive elimination reactions by providing the second redox equivalent through the alkyne complex ligand. In addition to setting suitable electrochemical potentials, the electronic cooperativity of the metals is of great importance for this purpose.
For these applications, we are looking for optimal combinations of metal ions and complex components that lead to desirable properties such as good solubility, configurational stability, redox variability in similar potential windows, high absorptivity and, last but not least, the presence of good spectroscopic probes such as CO ligands for IR spectroscopy or metals with suitable nuclear spin for EPR spectroscopy. The latter is fundamental for the elucidation of electronic structures in different redox states. The associated studies therefore include more specialized methods such as EPR spectroscopy, cyclic voltammetry, NIR, UV-vis and fluorescence spectroscopy, and spectroelectrochemistry, in addition to comprehensive routine characterization (X-ray diffraction, multinuclear NMR spectroscopy, IR/Raman spectroscopy).
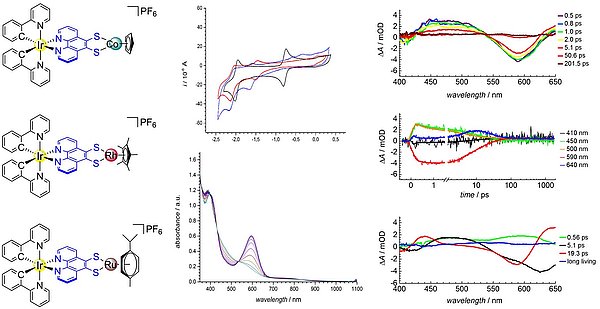
The work on light-induced charge separation and energy transfer in polynuclear complexes is based on studies with phenanthroline 5,6-dithiolate (phendt2-). Phenanthrolines with donor substitution in the 5,6-position are particularly rigid bridging ligands that fulfill an advantageous condition for very fast electron transfer processes between linked metals. In 2014, we presented the first dinuclear complexes with phendt2- combining N,N'-bound Ru(bpy)2- and Ir(ppy)2- photocenters with dithiolate-coordinated divalent group 10 metals. These studies were aimed at setting a thermodynamically favorable redox potential of the metals coordinated at the dithiolate moiety for photoinduced directional electron transfer. In all cases, coordination of a second metal at the dithiolate chelate results in effective photoluminescence quenching. Since 2014, a close collaboration exists with the group of Stefan Lochbrunner at the Department of Physics of the University of Rostock, Germany, which performed the time-resolved spectroscopic characterization of the complexes. For the compound [Ir(ppy)2(µ-phendt)Co(C5H5)], we were able to detect ultrafast Dexter energy transfer from the Ir complex to the CpCo unit using fs-transient absorption spectroscopy, which however could not be distinguished from a consecutive electron and hole transfer mechanism (see Figure). Consequently, extended phenanthroline ligands are ideal platforms for ultrafast electron transfer processes.
For more information, please visit our Uni-Hompage.
Literature:
Migratory insertion of isocyanide into a ketenyl–tungsten bond as key step in cyclization reactions C. Timmermann, P. Thiem, D. Wanitschke, M. Hüttenschmidt, J. Romischke, A. Villinger, W. W. Seidel, Chem. Sci. 2021, 10.1039/D1SC06149F
Metal/Metal Redox Isomerism Governed by Configuration S. Ludwig, K. Helmdach, M. Hüttenschmidt, E. Oberem, J. Rabeah, A. Villinger, R. Ludwig, W. W. Seidel, Chem. Eur. J. 2020, 26, 16811-16817.
Facile Synthesis of a stable side-on Phosphinyne Complex by Redox Driven Intramolecular Cyclisation H. Lange, H. Schröder, E. Oberem, A. Villinger, J. Rabeah, R. Ludwig, K. Neymeyr, W. W. Seidel, Chem. Eur. J. 2020, 26, 11492-11502.
Pathways to Polynuclear Complexes with α-S,N-Substituted Alkynes as Bridging Ligand J Rüger, C. Timmermann, A. Villinger, W. W. Seidel, Inorg. Chem. 2019, 58, 9270-9279.
1,10-Phenanthroline-dithiine iridium and ruthenium complexes: Synthesis, characterization and photocatalytic dihydrogen evolution E. Erdmann, A. Villinger, B. König, W. W. Seidel, Photochem. Photobiol. Sci. 2018, 17, 1056–1067.
Ultrafast Energy Transfer in Dinuclear Complexes with Bridging 1,10-Phenanthroline-5,6-Dithiolate E. Erdmann, M. Lütgens, S. Lochbrunner, W. W. Seidel, Inorg. Chem. 2018, 57, 4849–4863.
Sterically encumbered metalla-diphosphines: unlocking alkyne rotation by PtII coordination K. Helmdach, S. Dörk, A. Villinger, W. W. Seidel, Dalton Trans. 2017, 46, 11140-11144.
Synthesis and activation potential of an open shell diphosphine K. Helmdach, S. Ludwig, A. Villinger, D. Hollmann, J. Kösters, W. W. Seidel, Chem. Commun. 2017, 53, 5894–5897.
Minimalistic Ditopic Ligands: An α-S,N-Donor-Substituted Alkyne as Effective Intermetallic Conjugation Linker J. Rüger, C. Timmermann, A. Villinger, A. Hinz, D. Hollmann, W. W. Seidel, Chem. Eur. J. 2016, 22, 11191–11195.
Dual nucleophilic substitution at a W(II) η2-coordinated diiodo acetylene leading to an amidinium carbyne complex K. Helmdach, J. Rüger, A. Villinger, W. W. Seidel, Chem. Commun. 2016, 52, 2616–2619.